
In the pharmaceutical world, accurate documentation is the foundation of global drug approval. Every product that enters the market undergoes a rigorous review process, guided by the Common Technical Document (CTD) or its electronic version, eCTD.
For students pursuing careers in drug and regulatory affairs, understanding CTD and eCTD formats is essential. These documentation systems standardise how companies present data to regulatory authorities, ensuring consistency, clarity, and compliance across global markets.
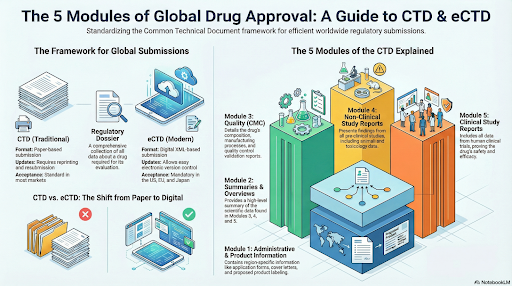
The Common Technical Document (CTD) was developed by the International Council for Harmonisation (ICH) to create a unified structure for regulatory submissions across the United States, Europe, and Japan. Before its implementation, every region followed a different submission format, leading to duplication and delays in drug approvals.
The CTD resolved this by offering a harmonised dossier structure that supports efficient review and communication between companies and regulatory bodies.
Takeaway: CTD ensures uniformity in how data is organised and submitted for global pharmaceutical approval.
A regulatory dossier is a comprehensive collection of scientific and administrative information about a drug. It includes every detail — from manufacturing methods and testing data to safety, efficacy, and labelling — required for product evaluation by authorities.
FAQ — Who prepares regulatory dossiers?
Regulatory Affairs (RA) professionals or documentation specialists prepare dossiers.
Their work involves compiling, reviewing, and submitting data to agencies like CDSCO,
USFDA, EMA, or MHRA for marketing authorisation.
Takeaway: Dossier preparation is one of the most vital responsibilities within the role of the drug regulatory affairs department.
The CTD provides a five-part structure that simplifies the submission process. It organises information logically, making it easier for reviewers to assess a product”™s quality, safety, and efficacy.
FAQ — What are the 5 modules of regulatory affairs?
The CTD is divided into five modules, each containing specific sections covering
administrative, quality, and clinical data.
| Module | Description | Contents |
|---|---|---|
| Module 1 – Administrative and Product Information | Contains region-specific information such as application forms and product labelling. | Cover letter, authorisation forms, labelling details. |
| Module 2 – Summaries and Overviews | Summarises quality, non-clinical, and clinical data. | Overall summaries of data in Modules 3–5. |
| Module 3 – Quality (Chemical, Pharmaceutical, Biological) | Details drug manufacturing, composition, and control. | Drug substance, drug product, validation reports. |
| Module 4 – Non-Clinical Study Reports | Presents pharmacology and toxicology findings from pre-clinical studies. | Animal test data, toxicity studies, safety analysis. |
| Module 5 – Clinical Study Reports | Includes clinical trial data on human subjects. | Bioavailability, efficacy, and post-marketing studies. |
Takeaway: Each CTD module builds upon the previous one — moving from administrative data to scientific validation — creating a complete product profile for regulatory review.
As technology evolved, the electronic Common Technical Document (eCTD) replaced paper-based CTD submissions. eCTD offers a more efficient and secure way to manage, update, and submit drug dossiers globally.
| Aspect | CTD | eCTD |
|---|---|---|
| Format | Paper-based submission | Digital XML-based submission |
| Accessibility | Physical documentation | Easily accessible via software platforms |
| Updates | Requires reprinting and resubmission | Allows electronic updates and version control |
| Compliance Tracking | Manual | Automated |
| Global Acceptance | Standard in most markets | Mandatory in the US, EU, and many Asian regions |
FAQ — Difference between CTD & eCTD?
CTD is a paper-based structure for regulatory submissions, while eCTD is its electronic
format that enables faster review, secure data exchange, and efficient version control.
Takeaway: eCTD is now the preferred format for global submissions due to its efficiency and traceability.
For students pursuing a career in drug and regulatory affairs , dossier preparation is a highly specialised skill that offers both domestic and international career opportunities.
FAQ — What is the job role in DRA documentation?
A documentation professional in Regulatory Affairs ensures that product data is
compiled, formatted, and submitted according to international guidelines, often working
with cross-functional teams in R&D, QA, and clinical departments.
Takeaway: Dossier preparation is a gateway to global pharmaceutical operations, offering career growth in regulatory consulting and documentation management.
Students aspiring to enter this domain can enhance their knowledge through structured programs such as:
These programs help learners understand dossier compilation, CTD/eCTD formatting, and global submission protocols. Those exploring cost-effective learning paths can research DRA course fees in Delhi to find suitable options.
Takeaway: Academic preparation through specialised diploma or certificate courses lays the foundation for a successful career in regulatory documentation.
The Common Technical Document (CTD) and its digital counterpart eCTD have transformed global regulatory submissions by introducing structure, efficiency, and transparency. Understanding their five modules is crucial for students aiming to work in documentation, compliance, or submission management.
In today”™s regulatory landscape, professionals trained in what is drug regulatory affairs are indispensable — they ensure that safe and effective medicines reach patients worldwide.
Final Takeaway: CTD and eCTD are not just documentation frameworks; they are the language of international drug approval — and mastering them opens doors to rewarding global careers.
Our counselling team is ready to assist you at every step — click the button below to get started.
Need advice? Our experts are on WhatsApp 24/7—message us anytime!